

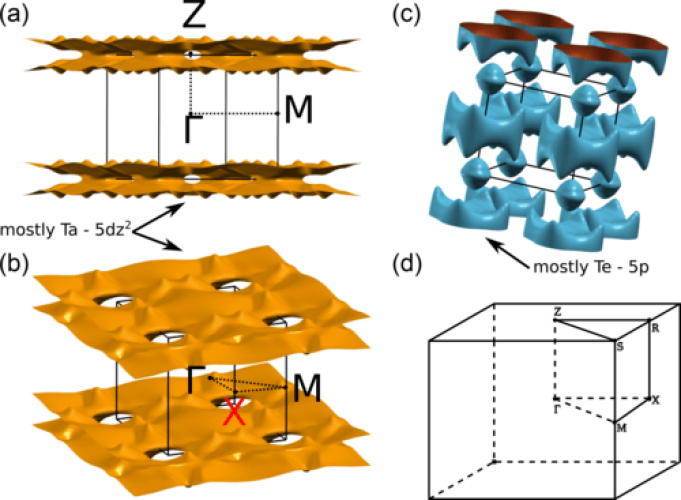
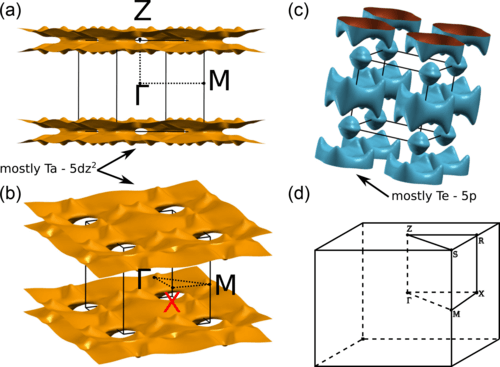
The origin of the charge density wave in TaTe4 is discussed on the basis of a first-principles density functional theory analysis of the Fermi surface, electron-hole response function, phonon band structure of the average structure, and structural optimization of the modulated phase. Analysis of the band structure and Fermi surface of the average structure clearly proves that despite the presence of TaTe4 chains in the crystal structure, TaTe4 is in fact a 3D material as far as the electronic structure near the Fermi level is concerned.
A Fermi surface nesting mechanism is dismissed as the origin of the 2a×2a×3c structural modulation. The optimized 2a×2a×3c structure, which is found to be the more stable modulation in agreement with the experimental observations, can be obtained directly from a soft-phonon mode computed for the undistorted structure. Our results suggest that the driving force for the distortion is the maximization of Ta-Ta metal-metal bonding subject to inducing the minimum bonding decrease in the Te sublattice.
Hits: 2095
M4ELC Sustainable Electronics
Competition between Ta-Ta and Te-Te bonding leading to the commensurate charge density wave in TaTe4
Bogdan Guster, Miguel Pruneda, Pablo Ordejón, and Enric Canadell
Phys. Rev. B 105, 064107 – Published 22 February 2022
DOI: https://doi.org/10.1103/PhysRevB.105.064107

